.svg)
.svg)
Complex injectables have become a popular delivery method in acute and chronic disease treatments for small molecules and biologicals. Particularly, suspensions and lipid-based nanoparticles are being increasingly utilized due to their ability to increase drug loading to improve bioavailability/stability and enable the formulation and development of long-acting injectables for poorly soluble drugs and biologicals.
Production of early phase clinical trial materials for complex injectables, however, is a challenge. Pharmaceutical and biotechnology companies can turn to a niche contract development and manufacturing organization (CDMO) to overcome the inherent obstacles. The right CDMO partner will have specialized technologies for complex injectable development along with the equipment, facilities and operations to adopt GMP practices with a “laboratory setting.” The best-in-class will also be nimble and have greater flexibility regarding changes, timing, and cost for successful manufacture of complex injectables in early phase development of therapeutic drugs.
Complex Injectable Dosage Form Development
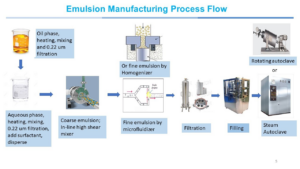 Complex parenteral products include delivery systems such as sterile suspensions, liposomes, lipid nanoparticles, emulsions, polymeric microspheres, and pellet implants. These are used to administer the drug via the injection, ophthalmic, subcutaneous, and implant routes.
Complex parenteral products include delivery systems such as sterile suspensions, liposomes, lipid nanoparticles, emulsions, polymeric microspheres, and pellet implants. These are used to administer the drug via the injection, ophthalmic, subcutaneous, and implant routes.
The scientific team on such a project needs to understand each delivery system:
Suspensions/nanosuspensions – The primary consideration for suspensions/nanosuspensions is they tend to settle over time and change in particle size distribution. This can be controlled by adding flocculating agents to enhance particle “dispersibility,” including viscosity enhancers to reduce the sedimentation rate in the flocculated suspension and selecting the appropriate stabilizer and surfactant.
Lipid-based nanoparticles – They are formed from lipids and other excipients in aqueous medium. For a lipid nanoparticle to be processed by 0.22-micron sterile filtration, its particle size distribution has to be well below 200 nm or an aseptic process needs to be utilized.
Emulsions/Microemulsions – Emulsions can be divided into three categories. Macroemulsions are typically opaque because the average particle size of the hydrophobic droplet is typically > 100 nm and thus scatters light. Microemulsions and nanoemulsions are obtained when the droplet size is typically <100 nm. Microemulsions are thermodynamically stable due to the use of sufficient co-solvents and co-surfactants to prevent Ostwald ripening. Nanoemulsions contain much less of the stabilizing co-solvents and co-surfactants, are meta-stable and more susceptible to Ostwald ripening.
Polymeric Nano/Microparticles – The drug can be protected from burst release and degradation with polymeric nano/macroparticles. This helps achieve prolonged drug delivery and a longer shelf-life.
GMP Considerations
When selecting a CDMO partner for complex injectables, GMP considerations must be taken into account, as well. This ensures the facility, personnel, and processes meet stringent guidelines and are in compliance.
Facility, Environment and Equipment – In early phase trials, the quantity of the drug substance is often extremely limited. Matching the equipment scale and material handling expertise with the product batch size is essential to ensure a successful, cost-effective outcome.
Companies that specialize in small-volume early-stage products, such as Ascendia, have significant experience in rapid small-scale manufacturing campaigns. A smaller support team generally has greater flexibility with regards to changes and timing. Larger CMOs tend to be more rigid and typically have defined systems that are not easily changed and create longer lead times.
The environment where aseptic processing occurs may require class 100 clean room suites, laminar flow hoods, biosafety cabinets, isolators, and restricted access barrier systems (RABSs). All air delivered to a cleanroom should pass through HEPA filters. The aseptic process should also include environmental monitoring to ensure microbiological control over the product.
Ascendia monitors its equipment used for sterilization with calibrated temperature probes and sterilization cycles. Our team documents and incorporates the findings into the batch record, as well. The product’s contact surface of manufacturing equipment requires cleaning verification following production. Items that cannot be sterilized must be disinfected before brought into the cleanrooms, and the entire area needs to be disinfected after processing.
Production Process and Validation – Our production process ensures the sterility and low endotoxin level of the product. If filtration is used for sterilization, filters are bubble point tested to certify integrity. To achieve the aim of a sterile product by an aseptic process, several aspects are considered and processes validated.
Process confirmation includes simulation tests using microbial growth media. The authentication covers filling of media, environmental monitoring, and incubation and evaluation of the filled vials. Microbial control is critical as part of cGMP for sterile products. Correct aseptic technique, properly functioning equipment, and adequate cleaning processes are demonstrated using media fills.
Quality Control – The Quality Control department releases the finished drug product per specification and review of the manufacturing batch record to ensure that procedures are followed, necessary investigations are performed, any subsequent corrective actions are incorporated. Such an approach assures that all testing performed during the process meets specifications so that the product is demonstrated to be sterile and meet product quality attributes.
Laboratory Controls – Ascendia calibrates and maintains laboratory equipment according to established written procedures. Personnel verify that the equipment is in good working condition when samples are analyzed. Initiation of a stability study using representative samples of the Phase 1 investigational drug to monitor the stability and quality of the Phase 1 investigational drug during the clinical trial are performed under ICH temperature, humidity, and light storage conditions.
Personnel and Training – Everyone at Ascendia involved in the manufacture of clinical trial material is trained in cGMP aseptic techniques. Our procedures are clearly written and followed throughout the project. For aseptic manufacturing processes, we provide and adhere to detailed standard operating procedures (SOPs). This includes aseptic operation, gowning, room cleaning, as well as personnel and room environmental monitoring procedures.

Contact us to learn more about Ascendia Pharma’s approach to overcoming the challenges of poorly soluble drugs. Let us show you how we can add value to your product portfolio and pipeline.
