.svg)
.svg)
Drug development teams are familiar with failure. After all, only one in 5,000 discovery compounds will reach the market, according to market data. Those odds can be increased if pharma companies select a contract development and manufacturing organization (CDMO) with experience – particularly in water-soluble drugs. The reason? Almost half of drugs on the market and nearly 90% of molecules in the discovery pipeline are poorly water-soluble. Leveraging advanced nanotechnologies is also critical to achieving success.
Most early development failures can be blamed on three issues – drug toxicity, safety, and efficacy. A significant increase in the percentage of new chemical entities (NCEs) with poor physical, chemical, and biopharmaceutical properties (BCS II and BCS IV) in the drug pipeline has played a significant role in attributing to those high failure rates.
Impact of Poor Solubility
Poor solubility can lead to low bioavailability, resulting in suboptimal drug delivery, ineffective drug efficacy, and side effects. As a result, various drug delivery nanotechnologies are critical in overcoming these bioavailability challenges faced by the pharma and biotech industries. Among the more successful are nano-suspensions, lipid microemulsions, nano-emulsions, and amorphous solid dispersions.
Teams developing immediate-release formulations must remember that there is typically a wide fluctuation of drug plasma concentration. The result is unwanted toxicity and poor efficiency. Compare this to oral controlled-release formulations that maintain a steady concentration of the drug in the plasma within the therapeutic index. For this reason, the latter formulations have been adapted for early drug development to overcome compound toxicity issues, as shown in figure 1.
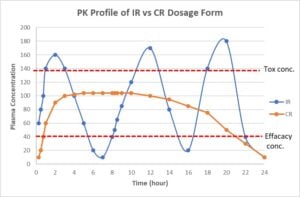
Figure 1
Controlled-release formulations can optimize the pharmacokinetic and pharmacodynamic properties of drugs, improving the safety and efficacy of NCEs that otherwise might fail for safety reasons. Another factor pharma and biopharma teams must consider is that controlled-release dosage forms have proven beneficial in lifecycle management of approved drugs via NDA 505(b)(1) or 505(b)(2) regulatory pathways. Reformulating multiple immediate-release daily doses into a once-a-day modified-release alternative simplifies dosing regimens, improves patient compliance, and enhances product safety.
CR Dosage Form Design Considerations
Every NCE has unique properties that require special considerations for rational design of a CR drug delivery system. CR dosage form development should define its target pharmaceutical profiles based on medical need, physiochemical/ biopharmaceutical properties, and applicability of formulation technology. Figure 2 shows the key considerations for design of CR dosage forms.
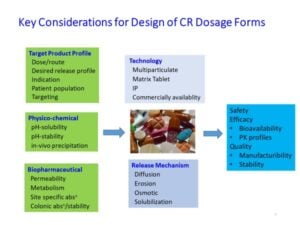
Figure 2
There are two main goals in early phase modified-release dosage form development. Enhancing efficacy, which is done by increasing the duration of Cmin plasma concentration above the effective concentration is one objective. The other is to minimize toxicity, achieved by reducing the Cmax plasma concentration to below the toxicity level. Controlled release can reduce the dosing frequency to improve patient compliance, as well.
The modified-release duration varies between 2 to 24 hours depending on the goals of the CR dosage form. Typical goals and associated release durations are:
1. If the CR formulation blunts the Cmax plasma concentrations to reduce Cmax-related side effects, a 4- to 6-hour delivery is sufficient
2. If a CR formulation improves efficacy by increasing duration of Cmin above the efficacious concentration and reduces dose frequency, a 6- to 24-hour release is desirable. (Note: the wide range is due to factors such as the drug body half-life and regional absorption of the drug in the lower GI tract)
3. If a CR formulation reduces local irritation or avoids drug degradation in the gastric fluid, a delay release of about 2 hours is satisfactory
When designing a CR dosage form, science teams at the pharma company and CDMO partner must collaborate to determine how to ease administration, dose accuracy, and swallowability. Specific patient populations, such as those suffering from Alzheimer’s disease as well as pediatric and geriatric segments, must have special consideration. Multi-particulate dosage forms, coated beads, and powder for reconstitution are popular for populations such as those due to ease of swallowing and reproducibility in drug-release rate.
Depending on body half-life, regional absorption in the GI tract, dose/solubility, bioavailability, matrix type, or membrane-coated CR dosage forms can be selected for development with an aid of modeling and simulation.
Solubility
For compounds with low water solubility, modifying release by controlling API particle size is not ideal. It is challenging to develop a CR dosage form for compounds with low solubility at a high dose level (dose/solubility >250). One way to resolve the issue is to use enabling formulation technologies, such as Ascendia’s NanoSol, AmorSol, and EmulSol nanotechnologies. They are proven to enhance API solubility and thereafter to incorporate the active soluble intermediate into a CR dosage form.
Stability
Some compounds are unstable in GI tract and subject to enzymatic degradation in the GI tract. Ascendia’s team of scientists are familiar with methods to stabilize the compound by using a buffer, protecting reagent and coating the API. A compound that is subject to colonic microflora digestion should not consider drug release in the lower GI tract with a release duration longer than ~6 hours.
Rational Design of CR Dosage Forms
The overall design and development process for controlled-release dosage forms of insoluble drugs can be divided into two steps:
1. To enhance the solubilities and dissolution rates as to the bioavailability of insoluble drugs
2. To incorporate the drug intermediate with enhanced solubility in an oral controlled-release dosage form, wherein the drug-release rate is controlled by the dosage form other than by the intrinsic low solubility of the API.
Figure 3 shows the decision tree for rational design of CR dosage form for fast translation of discovery compounds into the clinic.
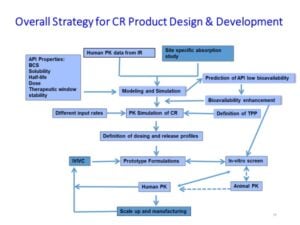
Figure 3
Through experience, our team has developed an understanding of key biopharmaceutical properties and how they relate to drug absorption and elimination. It has led to successful designs of CR dosage forms from discovery to first-in-human with a shorter timeline and lower development costs.
Contact us to learn more about Ascendia Pharma’s approach to overcoming the challenges of poorly water-soluble drugs. We can show you how we can add value to your product portfolio and pipeline.
