.svg)
.svg)
In looking at the current pharmaceutical market, there is a tremendous opportunity for drug developers when it comes to creating 505(b)(2) products. Part of it stems from the fact that more than half of the approved drugs presently available contain poorly water-soluble APIs. The common issues are poor bioavailability, suboptimal drug delivery, ineffective drug efficacy, and numerous side effects. For emerging pharma companies, partnering with the right contract development and manufacturing organization (CDMO) can help develop 505(b)(2) products more efficiently.
A 505(b)(2) new drug application (NDA) is one of three US FDA drug approval pathways. It has become a popular regulatory strategy, as submissions of 505(b)(2) applications, as well as the similar abbreviated new drug application (ANDA), have increased over the past few years.
Many drug developers must choose between the 505(b)2) and an ANDA. Using the FDA as guidance, an ANDA is for a duplicate of a previously approved drug that relies on the government agency’s finding that the reference listed drug (RLD) is safe and effective. There is also a petitioned ANDA, which is for a drug product whose dosage form, route of administration, strength, or active ingredient is different than an RLD.
ANDA differs somewhat from a 505(b)(2) application. The 505(b)2) is an NDA with complete reports on safety and effectiveness investigations. A portion of the information for approval is derived from studies not conducted by or for the drug applicant. A right of reference or use of those findings are not acquired by the applicant, either.
Benefits of 505(b)(2) Pathway
By this definition, manufacturers can travel the 505(b)(2) pathway to acquire FDA approval without performing all the work required with a traditional NDA. Such a strategy is an option to improve existing drug products with a new indication, dosage form, dosing regimen, strength, combination with other products, and new administration route. It can also be used to eliminate food effects, switch a drug from a prescription to over-the-counter, and orphan drug indications.1,2
Generic and brand companies are focusing on more complex 505(b)(2) products to separate themselves from commoditized generic competition. Many marketed drugs have been successfully reformulated to improve efficacy, safety, and patient compliance using the NDA 505(b)(2) or 505(b)(1) regulatory pathway (Table 1). Invigorating mature drug products using innovative drug delivery technologies or platforms can provide new marketing exclusivity and patent protection. It can help with product life cycle management, as well.
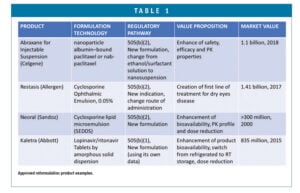
Table 1
Research Still To Do
Though pharma companies can save time and money with a 505(b)(2) pathway, it doesn’t eliminate all investigation. The CDMO-pharma company team must do their own research, particularly in chemistry, manufacturing, and control (CMC) factors or clinical areas required by FDA for approval that the product is safe, effective, and consistent between batches. 505(b)(2) drug development must completely understand the Drug Metabolism and Pharmacokinetics (DMPK), clinical pharmacology, biology, physical-chemical, and biopharmaceutical properties in relationship to drug dissolution, absorption, and the disposition process in the body.
Scientists must develop a rational formulation design with guidance from a decision tree to predict quantitative structure-activity relationships. In addition, understanding the advantages and limitations of drug delivery technologies to target pharmaceutical profiles, such as drug indication, dose regime, patient population, route of administration, and patent strategy (Table 2), are integral to a 505(b)(2) product being successfully developed the first time.
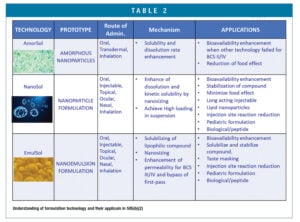
Table 2
Successful Nano Formulations
Ascendia Pharma has a suite of nanotechnology platforms, each of which has been used to advance 505(b)(2) drug products. Here are three examples:
ASD-002 – This platform is a ready-to-use, room temperature storage, nanoemulsion parenteral form of a blockbuster oral antiplatelet medicine for the treatment of Acute Coronary Syndrome. The current in-market drug, delivered orally, has a considerable delay when the medicine begins to take effect. In some cases, it can be several hours before the drug achieves peak concentration and therapeutic effect. Therefore, an injectable alternative is more ideal in time-sensitive environments, such as an acute, emergency setting. A few obstacles that need to be overcome to create this injectable version are the compound’s solubility, physical form, and chemical stability properties. Its nanoemulsion formulation allows ASD-002 to stabilize the compound and overcome these stability and delivery challenges.
ASD-004 – An ophthalmic nanoemulsion indicated to increase tear production, ASD-004 is particularly for patients whose tear production is presumed to be suppressed due to ocular inflammation associated with keratoconjunctivitis sicca. Dry-eye medications are only available as a white, opaque emulsion that are 100-200 nm in size, on average.
The efficacy of the leading dry-eye prescription medication is moderate (an estimated 15% according to prescribing information). Additionally, clinical trials revealed that the most common adverse reaction following the use of the drug was ocular burning (17%). Based on these factors, there is an unmet need to treat chronic dry-eye disease with a product that demonstrates improved response and fewer side effects. This can be achieved by reducing the level of surfactant and increasing formulation clarity.
Ascendia partnered with a pharmaceutical company to develop ASD-004, an optical-clear nanoemulsion eyedrop. Three potential advantages of the new formulation are:
- Droplet size below 100 nm – considerably smaller than the current alternative
- Nanoemulsion approach may eliminate side effect of blurred vision
- Possible reduction or elimination of the burning sensation due to lower surfactant level
ASD-005 – For sustained release of a non-selective β- and α-adrenergic receptor blocker by the parenteral route, Ascendia developed this liposome-like lipid nanoparticle. Current drugs are available only in immediate-release oral tablets twice daily and oral controlled-release once daily capsules. There is no parenteral dosage form in market.
Oral administration may present a challenge for patients under acute care conditions with congested heart failure or hypertension. The reason is that oral dosage forms typically have a delay in drug onset and extensive first-pass metabolism that results in an oral bioavailability of 25%-35%. Additionally, life-threatening hypotension and other side effects are frequently reported by patients. Therefore, a parenteral formulation with a rapid onset combined with a sustained-release characteristic is preferred for inpatients suffering from acute cardiovascular events.

Serving as a CDMO for a drug development company, Ascendia developed compositions containing carvedilol encapsulated in liposome-like nanoparticles that showed higher bioavailability and lower clearance rate. In vitro release of those liposomes in buffer solutions revealed extended release over 48 hours. In vivo animal data indicated that parenteral administration of drug encapsulated in lipid materials had a sustained-release PK profile.
To learn more and speak to a member of the Ascendia Pharma business development team, contact us directly at 732.640.0058.
References
