.svg)
.svg)
To meet the safety, efficacy, and quality standards of parenteral dosage forms, injectable solutions must be sterile and low pyrogen. They must also meet compendia specification requirements, including >90% label claim, related substance level lower than the tox qualified level, content uniformity, pH, osmolality, and particulate matter. They must be essentially free of visual foreign matter, as well.
Pharmaceutical teams developing injectable solutions should select a contract development manufacturing organization (CDMO) with extensive experience to ensure timelines are met and projects do not go over budget. CDMOs need to be familiar with three major challenges of parenteral dosage forms:
- Achievement of formulation stability
- Compatibility of drug substance with packaging components
- Sufficient drug concentration within a reasonable pH range and without using excipient levels that causes blood incompatibility and tissue irritation issues.
In this blog, we will discuss the stages of parenteral formulation development:
Preformulation Studies
Preformulation studies are critical for injectable solution development, which covers the measurement of solubility/stability as a function of pH, pKa, partition, moisture sorption/desorption, polymorphism and crystallinity. It does so by differential scanning calorimetry (DSC); thermogravimetric analysis (TGA) and powder X-ray diffraction; salt form screening; stability of drug substance; assessing API and excipient compatibility using stress testing; and forced degradation to evaluate the effect of pH, temperature, humidity, light, and oxidizing agents.
Compound solubility in commonly used solvent, cosolvent, surfactant, solubilizer, and complex agents are useful in setting the direction of formulation development, as well.
The aqueous solubility of a compound is dependent on its ionization state, including the ratio of ionized to unionized fraction. Ionization can be estimated using the Henderson–Hasselbach equation. For weak acidic compounds, (HA) is:
pH = pKa + log[A–]/[HA];
for weakly basic compounds, (BH) is:
pH = pKa + log[B]/[BH+],
where Ka is the ionization constant of the dissociation constant.
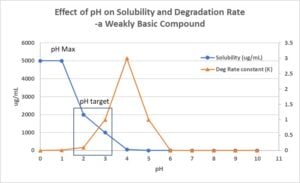
Figure 1
The effect of pH on API solubility and stability is most important for solution formulation development of ionizable compounds. It guides the CDMO when selecting the formulation pH in the solution dosage form (Figure 1).
Salt or co-crystal form is another factor that may also affect the compound solution solubility. Salt form screen or in situ salt formation should be evaluated to find a salt form that enhances compound solubility.
Formulation Development
Prototype formulation development covers selection of buffer, pH, stabilizers, isotonicity agents, antioxidants, preservatives, and solubility and/or viscosity-enhancing agents.
pH and Buffer Selection
pH is a critical aspect of parenteral formulation. The target pH should be as close as possible to physiological pH. The acceptable range is pH 2–11 for intravenous and intramuscular injection. It is pH 4-9 for subcutaneously injection due to potential irritation issue. A buffer, such as acetate, phosphate, citrate, histidine, and TRIS, can control the solution for an ideal range of pH 5-8. The buffer capacity should be kept to a minimum so body fluid can quickly adjust the formulation pH to physiological pH. If not, an irritation at the injection site may occur.
For IV infusion, the solution pH is typically controlled by the salt form of the drug or by acids and bases without buffer due to in vivo tolerability considerations. A compromise may be required between solubility and stability of the drug substance to find a target formulation pH where drug loading and product stability are acceptable.
Osmolarity
Parenteral formulations should be ideally isotonic. The most common agents to adjust the tonicity of a solution formulation are dextrose, Mannitol, glycerol, and sodium chloride. Hypertonic solutions cause blood cell deformation. A hypotonic solution leads to rupture of the blood cell and subsequent hemolysis. Formulations with an osmotic value 250 to 350 mOsm are acceptable; any exception to this range should be justified case by case.
Solubilizers
A high percentage of insoluble compounds are in the development stage. Therefore, solubilization is important for pharmaceutical scientists. In addition to altering pH and in situ salt formation, solubilizers or their combinations are routinely utilized to solubilize poorly water-soluble compounds. Co-solvents are used when a drug substance has insufficient solubility in a simple aqueous or pH-adjusted vehicle.
The water-soluble organic solvents and surfactants used in marketed injectables include propylene glycol, polyethylene glycol 300/400, glycerin, ethanol, polysorbate 80, Cremophor EL, N-methyl-2-pyrrolidone (NMP), etc. These solvents are usually combined using a pH adjustment. A decision tree can be used as a guide for the injectable solution development (Figure 2).
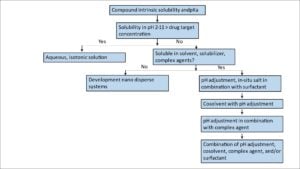
Figure 2.
Container and Closure
Compatibility of injectable solution with stopper and container is another critical aspect for drug development teams and their CDMO partners to consider. Potential discoloring, deformation, and leachable of the stopper due to incompatibility should be evaluated once the prototype formulation is chosen.
Leaching of alkaline materials from glass may cause an increase in solution pH. The reaction can be significant for un-buffered formulations, as the compound’s solubility and stability are sensitive to the pH change. CDMO teams evaluate container-closure integrity for each injectable solution during development before manufacturing and during stability study to ensure integrity of injectable products is maintained.
Process Development and Scale-up
Process development is centered around three critical areas for sterile products: sterility, stability, and particulate matter. Parenteral manufacturing requires filling and sealing in a Class 100 environment. For injectables processed under aseptic conditions, the manufacturing process should be validated using media runs to ensure sterility of the finished dosage forms.
Terminal sterilized products can be manufactured in a Class 10,000 environment. A sterilization cycle validation is necessary to ensure a 6-log reduction of bioburden. Critical process parameters that affect product potency and stability should also be evaluated during process development and scale-up.
Compatibility with Infusion Sets
IV formulations are typically injected by syringe and administered via infusion sets. The CDMO should evaluate if the compound is absorbed into the syringe and infusion set. Studies should indicate no major losses occur after filtration, injection, or infusion under the conditions of administration.
Plasma and Blood Compatibility
Compounds administered intravenously must be compatible with plasma and blood. Evaluation of the compounds in lead formulations should be undertaken initially in the blood and plasma of relevant preclinical species.
A plasma precipitation test should be conducted to reduce the risk of embolism during in vivo studies. The study is done by diluting the formulation with control plasma and watching for signs of precipitation. If the study reveals precipitation, an alternative formulation should be sought or the concentration of the formulation compound can be lowered.
The formulation compound should not cause hemolysis, significant crenellation, or erythrocyte clumping. There should also be no adverse reactions, such as thrombophlebitis, observed at the injection site following IV administration. A further evaluation in human blood and plasma should be undertaken in the formulation of choice for clinical trials.
Formulation Stability
A minimum storage life of 18 months, preferably at room temperature, is desired for a commercial parenteral solution formulation. If sufficient solution stability cannot be achieved by storing at room temperature, refrigerated storage at 2°C -8°C or at -20oC may be considered. Solution stability studies should be undertaken to predict storage life under both conditions.
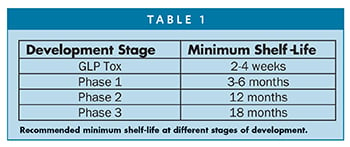
The recommended minimum shelf-life is different at various stages of development. Table 1 summarizes the minimum recommended shelf-life at different stage of development.
Summary
Injectable solutions are an attractive alternative to oral dosage form due to fast onset, reproducible PK/efficacy profile, high bioavailability, and suitability of administration under hospital setting. To overcome the associated obstacles, drug development teams should partner with a specialized CDMO experienced in developing formulations for injectable solutions.
Contact us to learn how we can be your CDMO for complex injectables for early phase studies.
This Formulation Forum was originally published on Drug Development & Delivery.

