.svg)
.svg)
Oral dosage drug forms represent a significant number of new drug approvals. In fact, they accounted for 45% (24 out of 53) of the new drugs given the green light by the FDA in 2020. Because of their place drug development, it’s important for pharmaceutical and biopharmaceutical companies to implement a phase-appropriate approach for early formulation development and later-stage commercial development.
Biotech companies prefer early phase development for their new compounds to prove the principle in human as soon as possible. Early phase formulation development typically uses “the simple formulation approach.” A sophisticated technology is necessary for compounds posing challenging properties in solubility and bioavailability.
By contrast, late-stage commercial formulation typically utilizes quality by design (QBD) principles and Design of Experiments (DoE). This approach helps develop a robust clinical formulation that is suitable for use as a commercial product, such as tablet, capsule, and liquid-filled capsule. Incorporating advanced technologies into regular tablet/capsules dosage forms is a daunting task for the late-stage development.
Even though the ideal situation is for a formulation to be used for early phase and the late-stage clinical development and New Drug Application (NDA) registration, it rarely occurs. Market-image formulation development is not initiated until Phase 2b for most of the clinical projects, because of tight budgets and timelines, as well as pressure to minimize development cost before proof of concept in human.
Developing Product Profiles that Meet QTTP and CQA Criteria
Drug development teams must understand multiple compound variables to meet Quality Target Product Profile (QTTP) and critical quality attribute (CQA) criteria, including clinical requirements on the systemic exposure and PK profiles, label claim, content uniformity, dissolution, and stability. To achieve this, a compound’s properties, indication, route of administration, disease model, patient population and dose range must be recognized. The result is a phase-appropriate formulation can be developed.
Per ICH Harmonized Tripartite Guideline: Q8(R2) Pharmaceutical Development, August 2009, the QTPP is “a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product.” The QTPP is an essential element of a QBD approach and forms the basis of pharmaceutical product design.
CQA for oral dosage forms typically includes identification, assay, content uniformity, dissolution, degradation products, residual solvents, water content, microbial limits, and container closure system. The identification of a CQA from the QTPP is based on the severity of patient harm should the product fall outside the acceptable range for that attribute. All quality attributes are target elements of the drug product and should be achieved through a good quality management system. Appropriate formulation and process design and development are integral, as well.
Early Phase Development Factors
The main goal for early phase development is to test the compound safety and efficacy for the intended therapeutic indications in animal and humans. For a simple formulation, such as solution/suspension, drug-in-capsule is always desirable for a fast transition to tox and human studies. A desired compound systemic exposure in testing subjects is a prerequisite to meet the goals of early development.
When working with compounds that have poor bioavailability, additional time and resources may have to be allocated to achieve a simple elegant dosage form that incorporates sophisticated formulation technologies. Not devoting enough of either may lead to the drug program having a higher risk of costly failure in first-in-man and during late stages. Recognizing this, Ascendia has established an in-house guideline for a rational approach of oral dosage form selection for compounds at different stages of development (figure 1).
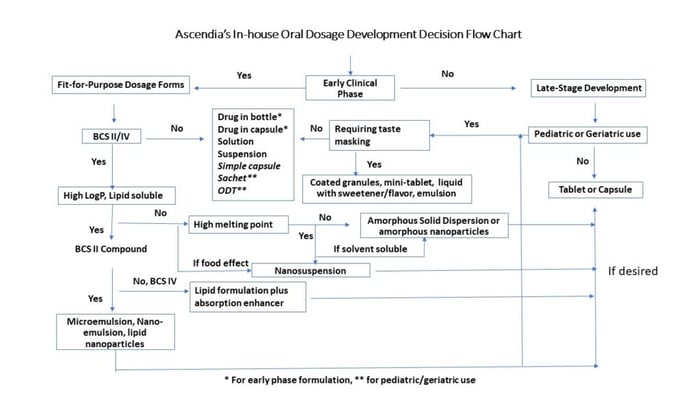
Various dosage forms can be selected for development with an aid of modeling and simulation. Factors to consider when making this decision are compound permeability, body clearance half-life, absorption in the GI tract, dose, pH solubility, therapeutic window and bioavailability. When designing a clinical dosage form, ease of administration, dose accuracy, and swallowability should be always considered for the target patient populations.
Early Phase Clinical Study Dosage Forms
Drug development teams usually consider four dosage forms during early phase clinical studies:
Drug-in-Bottle – The API compound is supplied in a bottle for constitution into solution or suspension. Commercial vehicles or their modified forms, such as “Ora-Sweet,” may be considered. Dosage can be further diluted into filled bottles for different dose levels, as well as for patients to take home.
Dose flexibility is the advantage of drug-in-bottle. Another benefit is a minimum stability requirement. This form is suitable for in-hospital dosing. In some cases, bulk amorphous solid dispersion formulations can be supplied in bottle and reconstituted.
Drug-in-Capsule – If a compound is readily wetted and dissolvable in GI fluids without excipients, drug-in-capsule can be considered. The active compound is filled in hard capsules. Drug-in-capsule is suitable for out-patient dosing and for studies requiring blinding.
Solution or Suspension – If the compound cannot be suspended or dissolved in a commonly used suspending vehicle but is stable for at least 3 to 6 months, a formulated solution, suspension, and nanosuspension filled in bottle at a CDMO can be considered. This dosage form is common for out-of-patient dosing. Lipidic formulation (SEDDS or Nanoemulsion) can be formulated and filled into bottle or hard capsules (as liquid-filled capsule) are considerations in certain scenarios.
Formulated Capsule and Tablet – For chronic dosing or a clinical program to fast progress to late-stage phase, formulated capsules or tablets may be desired. An API compound can be simply blended with a diluting excipient, and the blend is filled in hard capsules.
A formulated capsule or tablet may be considered for an API posing flowability, wettability, and dosage uniformity challenges. For poorly water-soluble compounds, enabling formulations can be further incorporated into the capsule and tablet dosage forms. Formulation examples include nanosuspensions, lipidic formulations, and amorphous solid dispersions. Liquid-filled capsules that contain lipidic formulation have been used for early- and late-stage development.
Late-stage Formulation Development Considerations
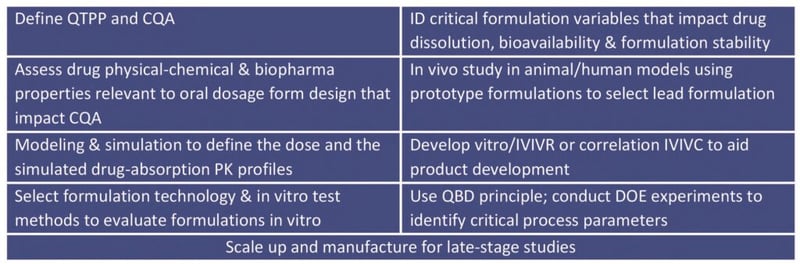
The table below lists the nine typical steps in oral formulation development.
Oral dosage forms for late stage and commercial production are preferably manufactured using conventional high-speed manufacturing processes. Traditional manufacturing process for oral dosage forms include:

Robust Formulation and Manufacturing Process
To achieve a robust formulation and manufacturing procedure, a CDMO must understand the critical processing parameters (CPPs) that impact CQA. For tablet or capsule development, API properties that can potentially impact content uniformity, dissolution, compression, stability and bioavailability are evaluated. Among the parameters to assess are granulation volume/time, blending speed and time, impeller and chopper speed, lubrication time, drying time/temperature, and compression force and speed.
For compounds with solubility and bioavailability hurdles, the overall design and development process for the late-stage dosage forms can be divided into the two steps. One is to enhance the solubilities and dissolution rates using an enabling technology. The second is to incorporate the drug intermediate with enhanced solubility in a traditional oral dosage form.
For formulation optimization, a DoE using factorial design may use 2-3 levels of critical excipients and their combination to evaluate their impact on CQA. An optimum level of the formulation composition, such as drug loading, API particle size, filler, binder, disintegrant, glidant, and lubricant of a tablet formulation, can be determined from the DoE experiment.
DoE experiments are conducted after the CPPs and CQAs have been selected based on risk assessment. This optimizes the process. A few process parameters with three levels in combination with API of different particle size evaluate the effects on granule flowability, compression, hardness, disintegration and dissolution. A design space is identified to define the control strategy for the large-scale manufacturing based on statistical analysis. Ascendia uses a Qualicap® F-40 liquid capsule filler in a solid oral cGMP manufacturing suite (figure 2). It has a speed of 40,000 unit/hour and is used for certain large-scale manufacturing projects.
Contact us to discuss how we can be your CDMO partner for oral dose drug formulation.
